Aus UroForum Heft 02/2025
Lothar Bergmann, Marit Ahrens
Nierenzellkarzinome (NZK) haben einen Anteil von ca. 85 % an Nierentumoren, wobei das klarzellige NZK mit einem Anteil von ca. 75–80 % die häufigste Entität ausmacht. Die nicht-klarzelligen Nierenzellkarzinome (nccRCC) haben einen Anteil von ca. 20–25 % und stellen histologisch und molekular mit über 20 Subentitäten eine heterogene Gruppe dar. Aufgrund der Seltenheit dieser Tumoren liegen nur unzureichende klinische Daten zur optimalen Therapiestrategie im fortgeschrittenen/metastasierten Stadium vor [1–3].
Pathologie der nccRCC
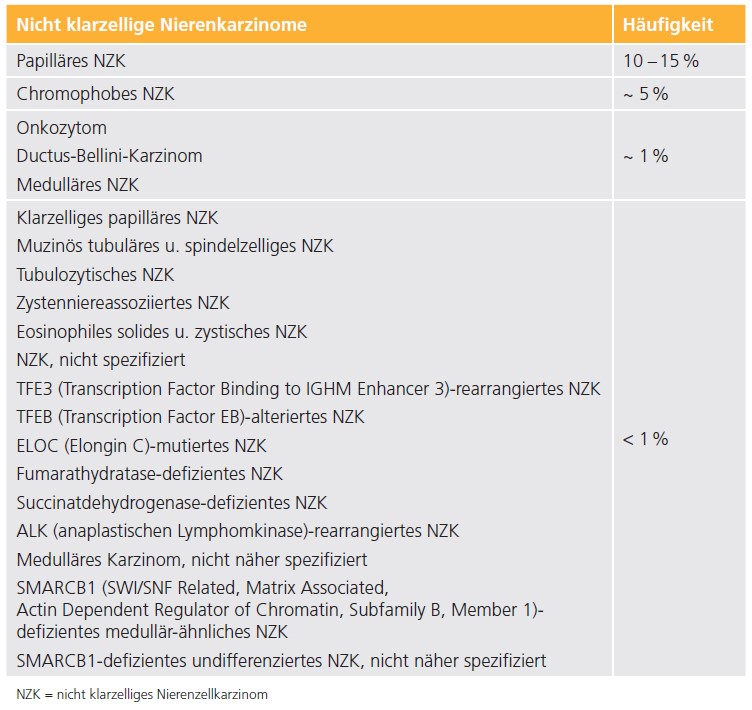
Die Klassifizierung der Nierentumoren erfolgt aufgrund morphologischer und molekularer Charakterisierung nach der WHO-Klassifikation 2022 [4]. Neben den klassischen histopathologischen Subtypen benigner Tumoren wie dem papillärem Adenom, Onkozytom und metanephrischem Adenom und malignen Tumoren wie dem konventionellen klarzelligen NZK, papillärem NZK, chromophobem NZK und Ductus-Bellini-RCC wurden viele neue RCC-Subtypen beschrieben (▶ Tab. 1) [4]. Das klarzellige NZK ist die häufigste Form des NZK und geht in etwa 85 % der sporadischen Fälle mit einer Alteration des Tumorsuppressorgens von Hippel-Lindau (VHL) einher. Beim zweithäufigsten NZK, dem papillären NZK, liegt häufig eine Alteration / Mutation mit Aktivierung des c-Met-Gens (MNNG HOS transforming gene) vor. Weiterhin wird nicht mehr in das papilläre NZK Typ I und Typ II unterschieden, da viele der Typ II papillären NZK anderen Entitäten zugeordnet werden konnten. Beim chromophoben NZK wurden Mutationen im Bereich der NADH (nicotinamide adenine dinucleotide) Dehydrogenase und der TERT (Telomerase reverse transcriptase) Promoterregion gefunden. Weitere Entitäten sind das Von-Hippel-Lindau-Syndrom (VHL) mit assoziiertem klarzelligen NZK, das Bird-Hogg-Dubé-Syndrom mit assoziierten hybriden, onkozytären Tumoren, Tuberose-Sklerose (TSC) mit assoziierten Angiomyolipomen oder PEComen sowie das eosinophile solide und zystische (ESC) NZK. Zu den familiären NZK gehören auch das SDH-defiziente (Succinat-Dehydrogenase defizientes) und das FH-defiziente (Fumarathydrogenase defizientes) NZK. Neue Tumorentitäten mit einer spezifischen molekularen Veränderung sind das „Transcription Elongation Factor 1“ (TCEB)-mutierte NZK, die MiTF-TFE-Familie von NZK (TFE3-Transloka- tions-assoziiertes, TFEB-transloziertes oder -amplifiziertes NZK), das SMARCB1 / INI-defiziente und das ALK-Translokations-assoziierte NZK. Letztere treten häufig bei jüngeren Patienten auf und haben oft einen besonders aggressiven Krankheitsverlauf mit dedifferenzierten, sarkomatoiden und rhabdoiden Merkmalen [2, 4, 5]. Nicht eindeutig klassifizierbare NZK werden mit der Diagnose NZK-NOS (not otherwise specified) klassifiziert.
Therapie des nccRCC
Lokalisierte nccRCC
Die Behandlung des nicht-metastasierten nccRCC erfolgt analog zu den klarzelligen NZK. In Abhängigkeit der Tumorgröße und -lokalisation kann eine Tumornephrektomie oder Nierenteilresektion durchgeführt werden. Bei fehlender rascher Progression sollte in der oligometastasierten Situation die Resektion der Metastasen erwogen werden [1, 6, 7].
Unklare Datenlage zur adjuvanten ICI-Therapie bei nccRCC
Daten zur Wertigkeit einer adjuvanten Therapie bei Patienten mit hohem Rezidivrisiko liegen beim nicht klarzelligen NZK nicht vor. Da die Zulassung in Europa unabhängig vom histologischen Subtyp für den Immuncheckpoint-Inhibitor (ICI) Pembrolizumab aufgrund der KN-564-Studie [6] besteht, kann die Durchführung einer adjuvanten Systemtherapie mit Pembrolizumab individuell diskutiert werden.
Metastasierte nccRCC
Patienten mit metastasiertem oder lokal fortgeschrittenem nicht klarzelligem NZK ohne Option zur R0-Resektion der Tumorlokalisationen sollen eine Systemtherapie erhalten [7,(8]. Gegenüber einer Monotherapie mit Tyrosinkinase-Inhi-bitoren (TKI) oder Inhibitoren des mammalian target of rapamycin-Signalwegs (mTOR) konnte durch eine Therapie mit ICI, ICI-Kombinationen (CTLA4-Antikörper plus PD- L1-Antikörper, z. B. Ipilimumab plus Nivolumab) oder TKI/ICI-Kombinationen eine deutliche Verbesserung des Gesamtüberlebens (OS) erreicht werden, wie mehrere randomisierte Studien besonders beim klarzelligen NZK zeigten (CheckMate-214, KEYNOTE-426, JAVELIN-101, CheckMate-9ER und CLEAR) [35–40]. Nicht klarzellige NZK wurden allerdings in den meisten randomisierten Studien mit ICI nicht eingeschlossen.
Bezüglich der Effektivität von TKI gibt es mehrere Studien, die einen Vorteil von Sunitinib gegenüber mTOR-Inhibitoren im progressionsfreien Überleben (PFS) oder OS bei nicht-klarzelligen NZK zeigten [16–18]. Lediglich bei chromophoben NZK zeigen mTOR-Inhibitoren eine gewisse Effektivität. Beim papillären NZK, das häufig mit einer c-Met-Aktivierung einhergeht, zeigte der TKI Cabozantinib, der auch c-Met blockiert, ein signifikant längeres PFS von 9 versus 5,6 Monaten mit Sunitinib, allerdings wurde kein Vorteil im Gesamtüberleben nachgewiesen [2, 19].
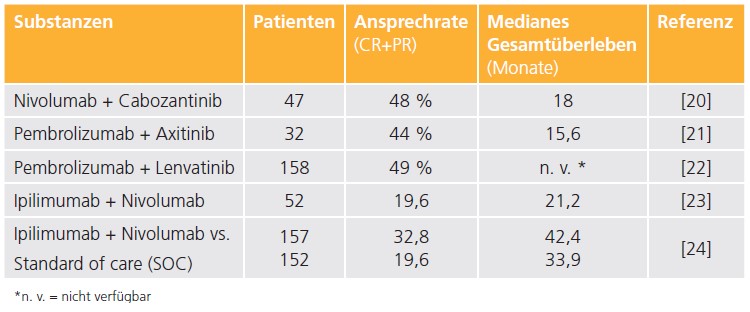
Eine einarmige Phase-II-Studie oder kleine randomisierte Studien mit ICI lassen vermuten, dass auch beim nicht klarzelligen NZK durch die Immuntherapie eine Verbesserung des Überlebens erreicht werden kann. In einer Phase-II-Studie mit Cabozantinib und Nivolumab lag das Gesamtansprechen bei 47,5 %, das PFS bei 12,5 Monaten und das mediane Gesamtüberleben bei 28 Monaten, wobei besonders papilläre NZK zu profitieren schienen [20]. Ähnliche Daten ergab eine Studie mit Pembrolizumab und Axitinib im Rahmen der einarmigen NEMESIS-Studie mit einem Gesamtansprechen von 44 % und einem medianen PFS von 10,8 Monaten [21]. In der KEYNOTE-B61- Studie zeigte die Kombination von Pembrolizumab mit Lenvatinib ein Gesamtansprechen von 49 % und ein medianes PFS von 18 Monaten. Die Überlebenswahrscheinlichkeit nach 12 Monaten lag bei 82 % [22]. Mit der Kombination Ipilimumab / Nivolumab wurde in der CheckMate-920-Studie ein Gesamtansprechen von 19,6 %, ein PFS von 3,7 Monaten und medianes OS von 21,2 Monaten erreicht. Patienten mit PD- L1-Expression sprachen besser an [23]. In einer kürzlich publizierten, randomisierten Studie mit Ipilimumab /Nivolumab versus Standard of Care (SOC; SUNNIFORECAST-Studie) bei 306 Patienten konnte ein Gesamtansprechen von 33 % vs. 20 % und eine signifikant verbesserte 12-Monats-Überlebensrate von 78 % vs. 68 % mit der ICI-Kombination gegenüber SOC (meistens TKI-Monotherapie) erreicht werden [24]. Besonders schienen hierbei Patienten mit einem CPS-Score (combined positive score) ≥ 1 zu profitieren. Eine Zusammenfassung der Studien beim nicht klarzelligen NZK mit ICI oder ICI/TKI-Kombinationen in der Erstlinie findet sich in ▶ Tabelle 2.
Die Europäischen Marktzulassungen für NZK erfolgten agnostisch für alle NZK subtypunabhängig, obwohl die evidenzbasierte Datenlage für klarzellige und nicht klarzellige NZK sehr unterschiedlich ist. Die nationale S3-Leitlinie in Deutschland empfiehlt daher eine Therapie der nicht klarzelligen NZK analog zu den klarzelligen NZK mit einer ICI / TKI-Kombination oder einer Immuntherapie-Dubletten-Behandlung in der ersten Linie. Mit Vorliegen der Daten der randomisierten SUNNIFORECAST-Studie liegt die höchste Evidenz jetzt für die Kombination Ipilimumab / Nivolumab vor. Die ESMO (European Society for Medical Oncology) empfiehlt aktuell Cabozantinib für die Erstlinienbehandlung des papillären NZK, einen TKI allein oder in Kombination mit einem mTOR-Inhibitor oder einem ICI für das chromophobe NZK, eine Immuntherapie-Kombination für das sarkomatoide NZK und eine platinbasierte Chemotherapie für Sammelrohr- und medulläre NZK [25]. Im Gegensatz dazu empfiehlt das amerikanische NCCN (National Comprehensive Cancer Network) Cabozantinib allein oder in Kombination mit einem ICI für jeden nccRCC-Subtyp außer für das Ductus-Bellini-Karzinom, für das eine platinbasierte Chemotherapie empfohlen wird [26].
Versuche, die den einzelnen Sub- typen zugrunde liegenden, genetischen und metabolischen Alterationen gezielt anzugreifen, waren bisher enttäuschend [2, 16]. Zusammenfassend können derzeit die nicht klarzelligen NZK analog zu den klarzelligen NZK therapiert werden, mit der derzeit höchsten Evidenz für Ipilimumab / Nivolumab aufgrund der SUNNIFORECAST-Studie. ◼
Interessenkonflikte:
L. Bergmann erklärt, dass bei der Erstellung des Beitrages keine Interessenkonflikte im Sinne der Empfehlungen des International Committee of Medical Journal Editors bestand.
M. Ahrens gibt Honorare für Advisory Board-Tätigkeiten von BMS, Janssen, Pfizer, Sanofi, MSD, Ipsen, Eisai, Amgen, AstraZeneca und Bayer an. Außerdem hat MA Kongresseinladungen von Ipsen, Merck, Pfizer sowie Reisekostenunterstützung durch BMS, Janssen, Pfizer, MSD, Ipsen, AstraZeneca und Bayer erhalten.
Literatur unter www.uroforum.de
Bildquelle: © Crystal light – stock.adobe.com

Korrespondenzadresse:
Prof. Dr. med. Lothar Bergmann
Schifferstraße 59
60594 Frankfurt/Main
Tel.: 069–905597822
onkologie@prof-bergmann.de