Nierenzellkarzinome (NZK) haben einen Anteil von ca. 85 % an Nierentumoren, wobei das klarzellige NZK mit einem Anteil von ca. 75–80 % die häufigste Entität ausmacht. Die nicht-klarzelligen Nierenzellkarzinome (nccRCC) haben einen Anteil von ca. 20–25 % und stellen histologisch und molekular mit über 20 Subentitäten eine heterogene Gruppe dar. Aufgrund der Seltenheit dieser Tumoren liegen nur unzureichende klinische Daten zur optimalen Therapiestrategie im fortgeschrittenen/metastasierten Stadium vor [1–3].
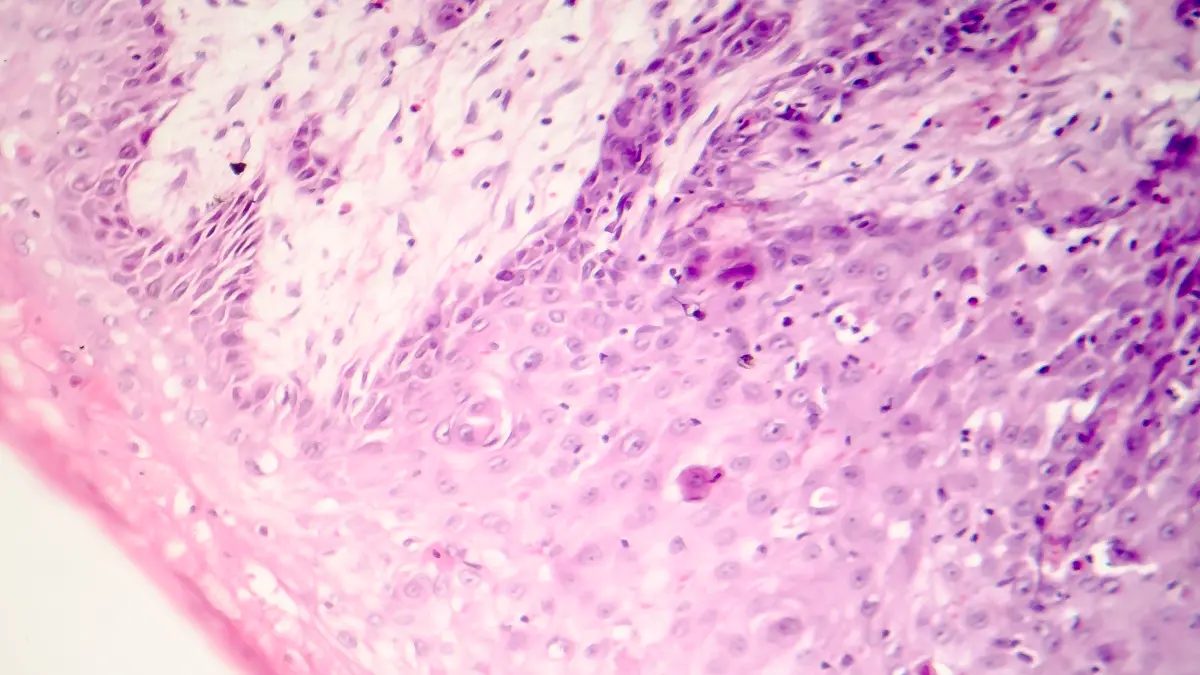
Pathologie der nccRCC Die Klassifizierung der Nierentumoren erfolgt aufgrund morphologischer und molekularer Charakterisierung nach der WHO-Klassifikation 2022 [4]. Neben den klassischen histopathologischen Subtypen benigner Tumoren wie dem papillärem Adenom, Onkozytom und metanephrischem Adenom und malignen Tumoren wie dem konventionellen klarzelligen NZK, papillärem NZK, chromophobem NZK und Ductus-Bellini-RCC wurden viele neue RCC-Subtypen beschrieben [4]. Das klarzellige NZK ist die häufigste Form des NZK und geht in etwa 85% der sporadischen Fälle mit einer Alteration des Tumorsuppressorgens von Hippel-Lindau (VHL) einher. Beim zweithäufigsten NZK, dem papillären NZK, liegt häufig eine Alteration/Mutation mit Aktivierung des c-Met-Gens (MNNG HOS transforming gene) vor. Weiterhin wird nicht mehr in das papilläre NZK Typ I und Typ II unterschieden, da viele der Typ II papillären NZK anderen Entitäten zugeordnet werden konnten. Beim chromophoben NZK wurden Mutationen im Bereich der NADH (nicotinamide adenine dinucleotide) Dehydrogenase und der TERT (Telomerase reverse transcriptase) Promoterregion gefunden. Weitere Entitäten sind das Von-Hippel-Lindau-Syndrom (VHL) mit assoziiertem klarzelligen NZK, das Bird-Hogg-DubéSyndrom mit assoziierten hybriden, onkozytären Tumoren, Tuberose-Sklerose (TSC) mit assoziierten Angiomyolipomen oder PEComen sowie das eosinophile solide und zystische (ESC) NZK. Zu den familiären NZK gehören auch das SDH-defiziente (Succinat-Dehydrogenase defizientes) und das FH-defiziente (Fumarathydrogenase defizientes) NZK. Neue Tumorentitäten mit einer spezifischen molekularen Veränderung sind das „Transcription Elongation Factor 1“ (TCEB)-mutierte NZK, die MiTFTFE-Familie von NZK (TFE3-Translokations-assoziiertes, TFEB-transloziertes oder -amplifiziertes NZK), das SMARCB1/INI-defiziente und das ALK-Translokations-assoziierte NZK. Letztere treten häufig bei jüngeren Patienten auf und haben oft einen besonders aggressiven Krankheitsverlauf mit dedifferenzierten, sarkomatoiden und rhabdoiden Merkmalen [2, 4, 5]. Nicht eindeutig klassifizierbare NZK werden mit der Diagnose NZK-NOS (not otherwise specified) klassifiziert.
Schlagworte zu diesem Beitrag
Ein Beitrag von
![]()
mgo medizin
Autor
Autor des Beitrags