Das Mantelzell-Lymphom (MCL) ist eine maligne Erkrankung des lymphatischen Systems und macht 5 – 7 % aller Non-Hodgkin-Lymphome in Nordamerika und Europa aus, mit medianem Erkrankungsalter von 60–70 Jahren. Auch wenn die Erkrankung historisch zu indolenten Lymphomen zählt, zeigt sie häufig einen aggressiven Verlauf mit medianen Überlebenszeiten von ca. 3 – 5 Jahren. In den letzten Jahren haben jedoch neue zielgerichtete Therapien die Behandlungsoptionen erheblich erweitert und zu einer deutlichen Verlängerung der Überlebenszeiten geführt [1].
Pathologische Merkmale, klinische Präsentation und Diagnostik
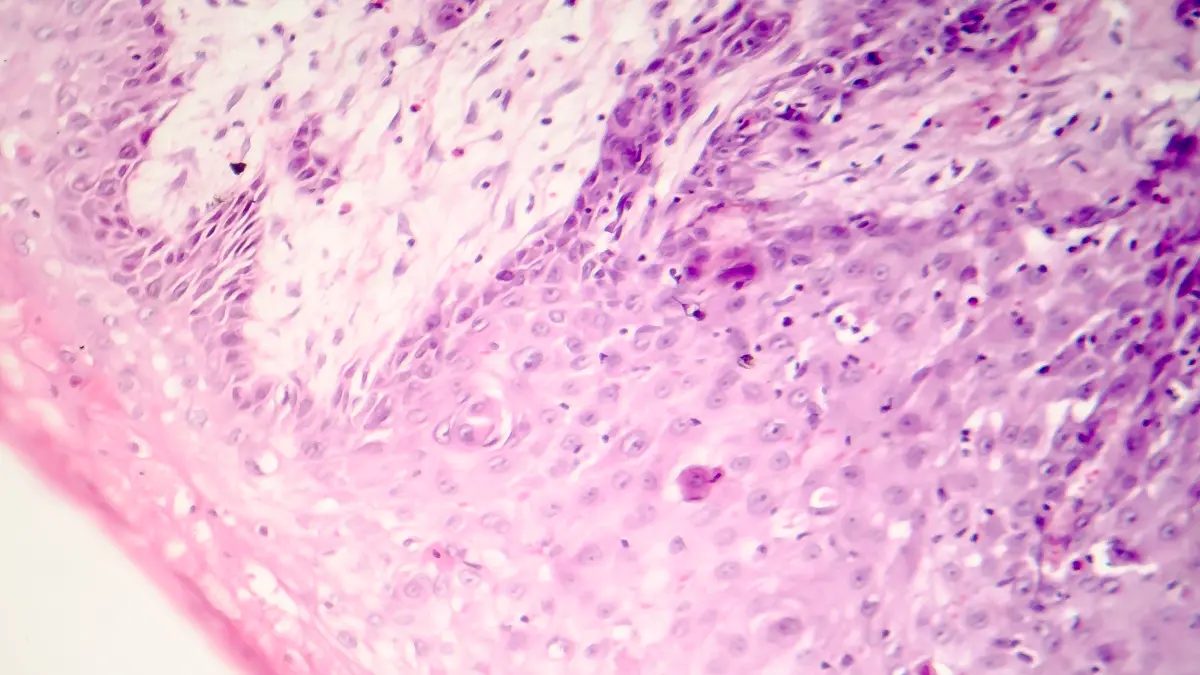
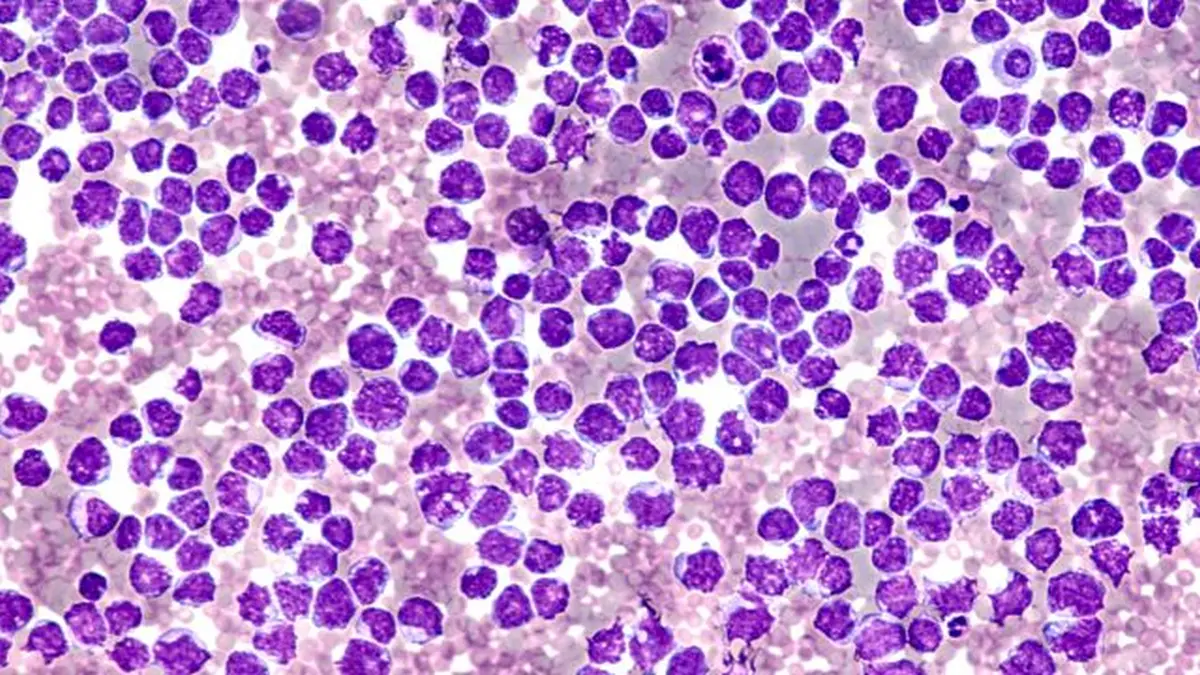
Das MCL geht von naiven B-Zellen der Mantelzone in Lymphfollikeln aus. Pathognomonisch ist die Translokation t(11;14)(q13;q32), die zur Überexpression von Cyclin D1 führt. In rund 95 % der Fälle lässt sich Cyclin D1 immunhistochemisch nachweisen; in seltenen Varianten werden alternativ Cyclin D2, D3 oder E exprimiert, meist begleitet von SOX11-Positivität. Weitere genetische Aberrationen – insbesondere TP53-Mutationen oder Deletionen von CDKN2A und ATM – sind häufig nachweisbar.
Klinisch imponiert MCL meist mit generalisierter Lymphadenopathie; rund 80 % der Patienten befinden sich bei Diagnosestellung im Stadium III / IV nach Ann-Arbor. B-Symptome sind seltener. Häufig bestehen Splenomegalie, Hepatomegalie sowie in 80–90 % der Fälle eine Knochenmarkinfiltration, teilweise mit leukämischer Ausschwemmung. Intestinale Manifestationen treten häufiger als bei anderen NHL auf. Diagnostisch entscheidend ist eine histologische Sicherung, bevorzugt durch komplette Lymphknotenexstirpation. In der Durchflusszytometrie zeigt typischerweise CD5+, CD20-bright Positivität sowie CD23- und CD200-Negativität. Differentialdiagnostisch abzugrenzen ist die chronisch lymphatische Leukämie (CLL), vor allem durch CD23 / CD200- Expression. Zur Initialdiagnostik gehören neben Anamnese, Labor und Lymphknotenhistologie auch Knochenmarkdiagnostik, Herz- / Lungenfunktionsprüfung, Hepatitis- / HIVSerologien und bildgebendes Staging (CT, ggf. FDG-PET-CT bei limitiertem Befall zur Planung einer Radiotherapie). Eine GI-Endoskopie sollte bei Verdacht auf intestinalen Befall erfolgen.
Gemäß WHO-Klassifikation werden klassische (nodale) und leukämischnon- nodale (SOX11-negative, meist indolente) MCL-Formen unterschieden sowie die In-situ-Mantelzellneoplasie (ISMCN) mit meist indolentem und inzidentellem Verlauf. Hochrisikofaktoren sind TP53-Mutationen, blastoide und pleiomorphe Morphologie und Ki-67 > 30 %. Das Outcome beim MCL lässt sich mit prädiktiven Mantle Cell Lymphoma International Prognostic Index (MIPI / MIPI-c) anhand von Alter, ECOG-Status, LDH, Leukozytenzahl und ggf. Ki-67-Index vorhersagen. Die Patienten können in Risikogruppen mit niedrigem, niedrig-mittelgradigem, hoch-mittelgradigem und hohem Risiko eingeteilt werden.
Bildquelle: David A. Litman – stock.adobe.com