Das hochaggressive Burkitt-Lymphom (BL) ist mit einer jährlichen Inzidenz von 2–3 Fällen pro 1 Million Einwohner in den Industrienationen eine seltene Erkrankung aus der Gruppe der Non- Hodgkin-Lymphome der B-Zell-Reihe, von denen es etwa 5 % der Erkrankungen ausmacht [1–3]. Man unterscheidet das endemische, oftmals Epstein-Barr-Virus (EBV)-assoziierte vom sporadischen, überwiegend EBV-negative BL, welches vor allem ältere, männliche Patienten betrifft. Darüber hinaus wird das BL gehäuft im Kontext der Immunoseneszenz bzw. der iatrogenen oder infektiologisch- vermittelten Immunsuppression beobachtet. Trotz oftmals guten initialen Ansprechens auf etablierte Polychemotherapie-Protokolle in Kombination mit dem CD20-gerichteten Antikörper Rituximab bestehen weiterhin offene Fragen zur Wahl des individuell passenden Therapieprotokolls, insbesondere unter Berücksichtigung der therapieassoziierten Nebenwirkungen, einer Beteiligung des zentralen Nervensystems (ZNS) sowie der Rezidivtherapie. Dieser Artikel beleuchtet die aktuellen Therapiemöglichkeiten des Burkitt-Lymphoms im Erwachsenenalter.
Diagnostik und molekulare Merkmale
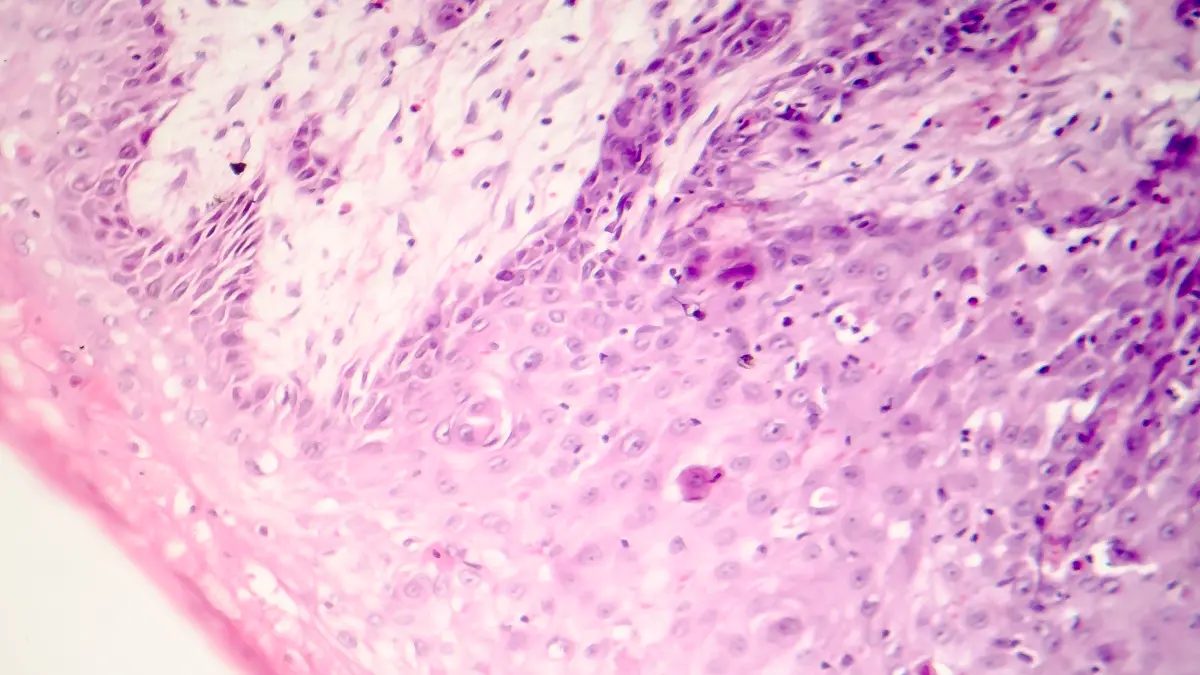
Die Diagnose des Burkitt-Lymphoms erfolgt nach initialer Verdachtsdiagnose, bei Symptomen wie B-Symptomatik, rapider Lymphknotenschwellung oder bildmorphologischer Verdachtsdiagnose, anhand histopathologischer sowie genetischer Untersuchungen einer repräsentativen Gewebeprobe. Hierfür ist initial häufig eine Lymphknotenextirpation, mindestens jedoch eine Stanzbiopsie, mit anschließender Begutachtung – im besten Fall aufgrund der teils schwierigen pathologischen Beurteilung durch eine Referenzpathologie – notwendig.
Das molekulare Kennzeichen des Burkitt-Lymphoms ist die charakteristische chromosomale Translokation t(8;14)(q24;q32), bei der das MYC-Protoonkogen auf Chromosom 8 in unmittelbare Nähe zum Locus der Immunglobulin-Schwerkette auf Chromosom 14 gebracht wird. Dies führt zu einer deregulierter MYC-Expression und infolgedessen zu einer unkontrollierten Zellproliferation. Dies zeigt sich charakteristischerweise häufig in einem sehr hohen KI67-Index (bis zu 100%), als zusätzlicher Marker des schnellen Tumorwachstums.
Mit der Überarbeitung der Klassifikation hämatologischer Neoplasien durch die Weltgesundheitsorganisation (WHO) im Jahr 2022 erfolgt die pathogenetische Einteilung aufgrund biologischer Krankheitsmerkmale in EBV-positive und -negative Burkitt-Lymphome. Zusätzlich wurde die Abgrenzung gegenüber anderen B-Zell-Neoplasien genauer beschrieben. Double-Hit-Lymphome werden nach neuer Klassifikation diagnostiziert, wenn sowohl eine MYC- als auch eine BCL2-Veränderung vorliegen. BCL6-Rearrangements werden nicht mehr berücksichtigt. Des Weiteren werden die vormals beschriebenen Burkitt-like Lymphome mit Aberration des Chromosoms 11q nun als hochgradiges B-Zell-Lymphom mit 11q-Aberration beschrieben [4, 5].
Zur Ausbreitungsdiagnostik sollte eine Bildgebung, präferiert mittels PET-CT, erfolgen. Die Durchführung des PET-CTs sollte den Therapiestart jedoch nicht maßgeblich verzögern. Alternativ können andere bildgebende Verfahren (CT/MRT) verwendet werden. Häufig finden sich BLManifestationen im zentralen Nervensystem (ZNS) extranodulär, z.B. als abdomineller Bulk, sowie im Knochenmark. Neben einer diagnostischen Liquorpunktion ist nach individueller Abwägung ggf. auch ein MRT des ZNS zu berücksichtigen. Die Stadieneinteilung erfolgt bei erwachsenen Patienten nach Ann- Arbor, bei pädiatrischen Patienten nach der St.-Jude-Klassifikation. Unabhängig des Stadiums erfolgt die Therapie mittels Polychemotherapie, wobei – vor allem bei pädiatrischen Patienten – die Intensität der Chemotherapie durch das Ausbreitungsstadium beeinflusst wird.
Schlagworte zu diesem Beitrag
Ein Beitrag von
![]()
mgo medizin
Autor
Autor des Beitrags