Die Transthyretin-Amyloidose (ATTR) ist eine fortschreitende, systemische Erkrankung, die durch die Ablagerung fehlgefalteter Transthyretin-Proteine (TTR) in verschiedenen Organen gekennzeichnet ist. Je nach genetischer Form, hereditär (ATTRv) oder Wildtyp (ATTRwt), und individueller Krankheitsausprägung kann die Symptomatik sehr unterschiedlich sein [1].
Früh im Krankheitsverlauf stehen bei vielen Patientinnen und Patienten neurologische Beschwerden im Vordergrund, insbesondere in Form sensomotorischer Polyneuropathien, vegetativer Dysfunktionen oder Karpaltunnelsyndromen [2]. Diese Beschwerden führen häufig zunächst in die neurologische Sprechstunde. Demgegenüber bleibt die kardiale Beteiligung, die für den Krankheitsverlauf und die Prognose jedoch von zentraler Bedeutung ist, häufig über längere Zeit unerkannt oder wird fälschlich anderen, häufigeren Krankheitsbildern wie hypertensiver Herzerkrankung, diastolischer Herzinsuffizienz oder hypertropher Kardiomyopathie zugeschrieben.
Ein solcher diagnostischer Irrweg kann wertvolle Zeit kosten, da sich gerade bei kardialer Manifestation die Prognose bei ausbleibender Behandlung deutlich verschlechtert. Für Neurologinnen und Neurologen ergibt sich hier eine Schlüsselrolle: Durch die initiale Vorstellung der Patientinnen und Patienten mit peripher-neurologischen oder autonomen Symptomen bietet sich die Möglichkeit, über ein gezieltes Erkennen begleitender kardialer Hinweise eine frühzeitige Verdachtsdiagnose und die Weichen für eine interdisziplinäre Abklärung zu stellen. Ein geschärfter klinischer Blick für die systemische Natur der ATTR-Erkrankung, jenseits der neurologischen Manifestationen, kann somit entscheidend dazu beitragen, kardiale Verläufe rechtzeitig zu erkennen und die heute verfügbaren krankheitsmodifizierenden Therapien optimal zur Anwendung zu bringen.
Frühe Hinweise auf eine kardiale ATTR
Typische neurologische Erstsymptome bei der varianten ATTR (ATTRv) oder auch bei der Wildtyp-Form (ATTRwt) sind distal betonte Dysästhesien, Gangunsicherheit, autonome Symptome wie orthostatische Hypotonie oder gastrointestinale Dysregulation. Diese Symptome führen häufig zu einer Vorstellung in der Neurologie und bieten somit eine entscheidende Chance zur Erkennung kardialer Komorbidität.
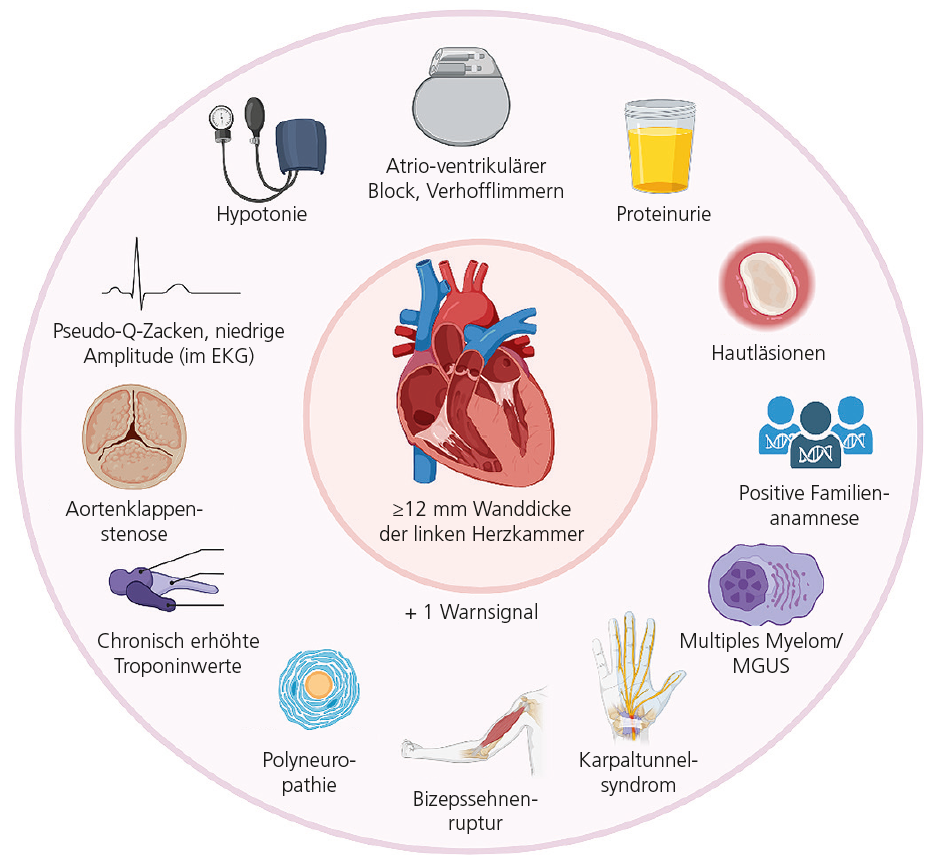
Folgende Red Flags weisen auf eine kardiale Beteiligung hin, und sollten bei neurologischen Patientinnen und Patienten gezielt erfragt oder untersucht werden (▶ Abb. 1):
- Belastungsdyspnoe oder eingeschränkte Belastbarkeit ohne klare pulmonale Ursache
- Unklare Synkopen oder Schwindel bei Positionswechsel
- Herzrhythmusstörungen (v. a. Vorhofflimmern)
- Niedervoltage im EKG trotz linksventrikulärer Hypertrophie im Echo
- Bilaterales Karpaltunnelsyndrom in der Anamnese
Diese Befunde weisen auf eine Amyloid-Kardiomyopathie hin, die bei ATTR-Patientinnen und Patienten häufig zunächst „stumme“ kardiale Veränderungen zeigt, bevor sich manifeste Symptome entwickeln [1].
Die Rolle der Neurologie im interdisziplinären Setting
Neurologinnen und Neurologen nehmen eine zentrale Rolle in der Diagnostik und Früherkennung der ATTR ein, insbesondere, wenn sich die Erkrankung initial durch sensomotorische oder autonome Symptome äußert. In vielen Fällen suchen Patientinnen und Patienten mit diffuser Polyneuropathie, Gangunsicherheit, Karpaltunnelsyndromen oder orthostatischer Dysregulation zunächst eine neurologische Praxis auf. Diese erste Anlaufstelle bietet eine entscheidende Gelegenheit, über den rein neurologischen Befund hinaus auf eine mögliche Beteiligung des Herzens zu achten. In spezialisierten Amyloidosezentren zeigt sich immer wieder, dass die Diagnose häufig auf neurologischem Wege angestoßen wird, während sich kardiale Manifestationen, wie diastolische Dysfunktion, Arrhythmien oder unklare Herzinsuffizienzsymptome, oft erst im weiteren Verlauf manifestieren oder retrospektiv als Teil der initialen Symptomatik erkannt werden. Ein vorausschauendes Denken in und systematische Erfassung von Symptomen und Red Flags ist daher essenziell.
Die enge interdisziplinäre Zusammenarbeit mit der Kardiologie, Hämatologie, Humangenetik und Nuklearmedizin ist dabei unerlässlich. Während die Neurologie häufig den diagnostischen Impuls liefert, erfolgt die weiterführende Abklärung, etwa durch kardiale Bildgebung, Biomarker-Analysen und genetische Testung. Diese sektorübergreifende Kooperation ermöglicht eine rasche Diagnosesicherung und einen frühzeitigen Therapiebeginn, der angesichts der progressiven Natur der ATTR-Erkrankung entscheidend für den langfristigen Verlauf ist.
Für die neurologische Praxis bedeutet dies: Das Bewusstsein für ATTR sollte bei unklaren Polyneuropathien, autonomen Symptomen oder bei einer entsprechenden Familienanamnese geschärft sein und ergänzt werden durch gezielte Rückfragen zu kardialen Beschwerden, Synkopen oder Rhythmusstörungen. Nur wenn die Muster („pATTRns“) erkannt werden, kann der Weg in eine zielgerichtete, multidisziplinäre Diagnostik und Therapie geebnet werden.
Empfohlene diagnostische Schritte bei Verdacht auf ATTR-Amyloidose mit kardialer Beteiligung [1]:
- Klinische Abklärung neurologischer und vegetativer Symptome
- Ruhe-EKG und Echokardiographie (v. a. diastolische Funktion, Wanddicke, Strain-Muster)
- Labor (NT-proBNP, Troponin) als Screeningparameter
- DPD-Skelettszintigraphie zur Detektion kardialer Amyloidablagerungen
- TTR-Genanalyse zur Differenzierung zwischen ATTRv und ATTRwt
Therapeutische Implikationen: Zeit ist Herz
Die kardiale Beteiligung bestimmt wesentlich die Prognose der ATTR-Amyloidose, sowohl bei der ATTRv als auch bei der ATTRwt. Eine frühzeitige Diagnose ist entscheidend, da mittlerweile mehrere krankheitsmodifizierende Therapien zur Verfügung stehen [3], die das Fortschreiten der Erkrankung signifikant verlangsamen und die Lebensqualität verbessern können. Zentraler Bestandteil der medikamentösen Therapie sind Stabilisatoren und Silencer. Tansthyretin-Stabilisatoren stabilisieren TTR-Tetramere in Ihrer physiologischen Form, und verhindern so die Bildung von Amyloidfibrillen. Der erste zugelassene Wirkstoff in dieser Klasse ist Tafamidis, der sowohl bei ATTRv als auch bei ATTRwt mit kardialer Beteiligung eingesetzt wird und in klinischen Studien eine signifikante Reduktion der Mortalität und Hospitalisierungsrate gezeigt hat [4]. Eine neue Alternative ist Acoramidis, ein weiterer TTR-Stabilisator, der kürzlich ebenfalls die Zulassung erhalten hat und sich in klinischen Studien durch eine starke TTR-Stabilisierung und eine gute Verträglichkeit auszeichnet [5]. Eine zusätzliche therapeutische Option bietet der RN
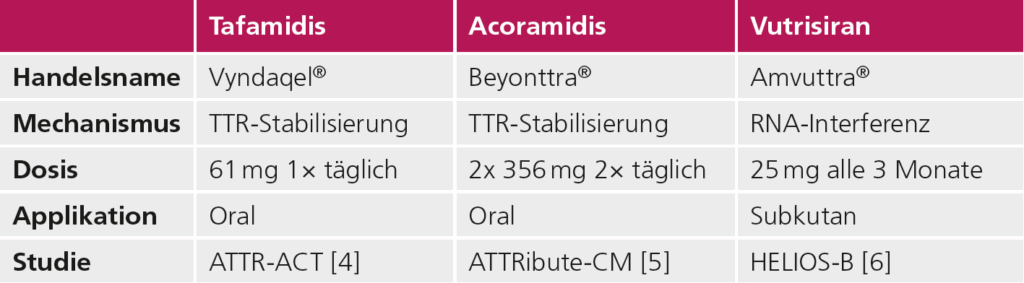
Wichtig zu beachten ist, dass klassische Herzinsuffizienzmedikamente, wie ACE-Hemmer, AT1-Blocker oder Betablocker bei Patientinnen und Patienten mit ATTR-CM häufig schlecht vertragen werden, insbesondere aufgrund von Hypotonien oder bradykarden Rhythmusstörungen, was die therapeutische Steuerung zusätzlich erschwert [7]. Die Auswahl der Therapie sollte individuell unter Berücksichtigung der klinischen Manifestation, genetischen Form, Organbeteiligung und Begleiterkrankungen erfolgen. Umso wichtiger ist es, die Erkrankung früh zu erkennen. Hier kommt der Neurologie eine Schlüsselrolle zu, insbesondere in der initialen Abklärung unspezifischer neuropathischer oder autonomen Beschwerden. Nur im interdisziplinären Zusammenspiel mit Kardiologie, Genetik und Nuklearmedizin kann eine zielgerichtete und effektive Therapie rechtzeitig eingeleitet werden.
Fazit
Bei unklaren Polyneuropathien oder vegetativen Symptomen sollte immer auch an eine potenzielle systemische Amyloidose mit kardialer Beteiligung gedacht werden. Besonders in frühen Krankheitsstadien liegt der Schlüssel zur Diagnose häufig in der neurologischen Anamnese und Befundkonstellation, schon bevor sich manifeste kardiologische Beschwerden zeigen. Ein interdisziplinärer Blick auf EKG, Echokardiographie und Laborwerte kann den entscheidenden Hinweis geben, und den Weg für eine zielgerichtete Therapie ebnen. Neurologische Praxen und Kliniken sind daher von zentraler Bedeutung für das frühzeitige Erkennen der ATTR-Erkrankung, insbesondere, wenn typische Red Flags wie autonome Dysfunktion, Karpaltunnelsyndrome oder sensomotorische Beschwerden in Kombination mit unklaren kardiovaskulären Symptomen auftreten. Nur wenn wir die „pATTRns“ erkennen, können wir Patientinnen und Patienten rechtzeitig und effektiv behandeln und ihnen damit nicht nur eine präzisere Diagnostik, sondern auch Zugang zu krankheitsmodifizierenden Therapien ermöglichen, die die Prognose und Lebensqualität nachhaltig verbessern können. Ein geschulter Blick, vernetztes Denken und frühzeitige interdisziplinäre Zusammenarbeit sind dafür essenziell.
Literatur
1. Garcia-Pavia P et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021; 42(16):1554–68
2. Conceição I et al. „Red-flag“ symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst 2016; 21(1): 5–9
3. Vogel J et al. Current Therapies and Future Horizons in Cardiac Amyloidosis Treatment. Curr Heart Fail Rep 2024
4. Maurer MS et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018; 379(11): 1007–16
5. Gillmore JD et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2024; 390(2): 132–42
6. Fontana M et al. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N Engl J Med 2025; 392(1): 33–44
7. Aus dem Siepen F et al. Standard heart failure medication in cardiac transthyretin amyloidosis: useful or harmful? Amyloid 2017; 24(sup1): 132–3
Interessenskonflikte
J. Vogel erklärt, persönliche Honorare außerhalb der eingereichten Arbeit von Eli Lilly, Bayer, Alnylam und Pfizer erhalten zu haben sowie die Forschungsförderung „UMEA Clinician Scientist Stipendium“ der Medizinischen Fakultät der Universität Duisburg-Essen sowie Bayer für eine gesponserte gemeinsame Studie außerhalb der eingereichten Arbeit. L. Michel erklärt, persönliche Honorare außerhalb der eingereichten Arbeit von Bayer, Bristol Myers Squibb, Alnylam, AstraZeneca, IFFM e. V. und vom Bund der Niedergelassenen Kardiologen (BNK) erhalten zu haben sowie als Forschungsförderung „IFORES Clinician Scientist Stipendium“ der Medizinischen Fakultät der Universität Duisburg-Essen sowie Bayer für eine gesponserte gemeinsame Studie außerhalb der eingereichten Arbeit.
© mgo fachverlage, all rights reserved
Korrespondenzadressen
Dr. med. Julia Vogel
Klinik für Kardiologie und Angiologie, Westdeutsches Herz- und Gefäßzentrum
Westdeutsches Amyloidosezentrum
Universitätsklinikum Essen
Hufelandstraße 55
45147 Essen
PD Dr. med. Lars Michel
FESC, Klinik für Kardiologie und Angiologie, Westdeutsches Herz- und Gefäßzentrum
Universitätsklinikum Essen
Hufelandstraße 55
45147 Essen
Tel.: +49 201 72384841
lars.michel@uk-essen.de