Forschende wiesen die erfolgreiche Wirksamkeit einer Gentherapie gegen Erblindung bei der seltenen Kinderdemenz »CLN2-Batten-Syndrom« in Netzhaut-Organoiden und Retina-on-Chip-Modellen nach. Eine neu entwickelte Gentherapie konnte die krankheitsbedingten Prozesse mindern und kann potenziell das Fortschreiten der Erblindung verhindern. Dies ebnete bereits den Weg für eine klinische Studie.
Krankheitsmechanismen
Die neuronale Ceroid-Lipofuszinose Typ 2 (CLN2) ist eine sehr seltene erbliche Kinderdemenz, verursacht durch Mutationen im sogenannten Tripeptidyl-peptidase-1(TPP1)-Gen. Diese Mutation führt zu einer Abwesenheit oder starken Reduktion des TPP1-Proteins, dieser Mangel wiederum stört den Proteinabbau und führt zu einer lysosomalen Ansammlung von Lipofuszinen in Gehirn, Netzhaut und anderen Organen. Dadurch kommt es bereits ab dem Kleinkindalter zu fortschreitender Degeneration dieser Organe. Besonders betroffen davon sind die Fotorezeptoren. Betroffene Kinder erblinden innerhalb weniger Jahre und zeigen unter anderem auch Symptome wie Krampfanfälle und Sprachentwicklungsstörungen. Ohne Behandlung führt die Krankheit in der Regel im mittleren Teenageralter zum Tod. Die Standardtherapie mit Enzym-Replacement-Therapie kann nur das Fortschreiten einiger Krankheitssymptome verlangsamen, aber es gibt keine Behandlung für den Verlust des Sehvermögens.
Humane Organoide und Organ-on-Chip-Modell
In Tiermodellen konnte die äußere Retinadegeneration nicht repliziert werden, daher wurde nach anderen Modellen zur Therapie des Sehverlusts gesucht. Einem Tübinger Forschungsteam vom Institut für Neuroanatomie & Entwicklungsbiologie und dem Institut für Biomedical Engineering ist es nun gelungen, in einem komplexen Modell der menschlichen Netzhaut die Erkrankung realitätsgetreu zu simulieren. Dies geschah in sogenannten Retina-Organoiden aus Stammzellen in Kombination mit einem »Retina-on-Chip«-Modell. In diesem Plastikchip ließen sich die Strukturen und Funktionen der menschlichen Netzhaut so realitätsnah abbilden, dass sich die bei CLN2 krankheitstypische Lipofuszin-Ablagerung präzise nachweisen ließ. Im Chip wird die natürliche Mikroumgebung der Netzhautzellen detailgetreu nachgebildet und eine Art künstlicher Blutfluss ermöglicht. Dadurch konnten präklinische pharmakologische Untersuchungen und eine Validierung einer Gentherapie an diesem Modell durchgeführt werden.
Gentherapie stoppt Ablagerungen im Modell
In diesem In-vitro-Krankheitsmodell wurde nun eine von Tern Therapeutics entwickelte Gentherapie (TTX 381) getestet. Ein modifiziertes Virus dient dabei als Transportvehikel. Ähnlich einem kleinen Shuttle überträgt es eine funktionierende Kopie des fehlenden TPP1-Gens präzise in die Netzhautzellen. Der sogenannte Adeno-assoziierte Virusvektor ist dabei kein Krankheitsüberträger und wird lediglich als eine Art leere Hülle zum Einschleusen des fehlenden Gens genutzt. Die Ergebnisse zeigen, dass die Produktion des fehlenden Enzyms wiederhergestellt werden konnte. Die schädlichen Lipofuszinablagerungen gingen durch die Behandlung zurück oder traten erst gar nicht auf.
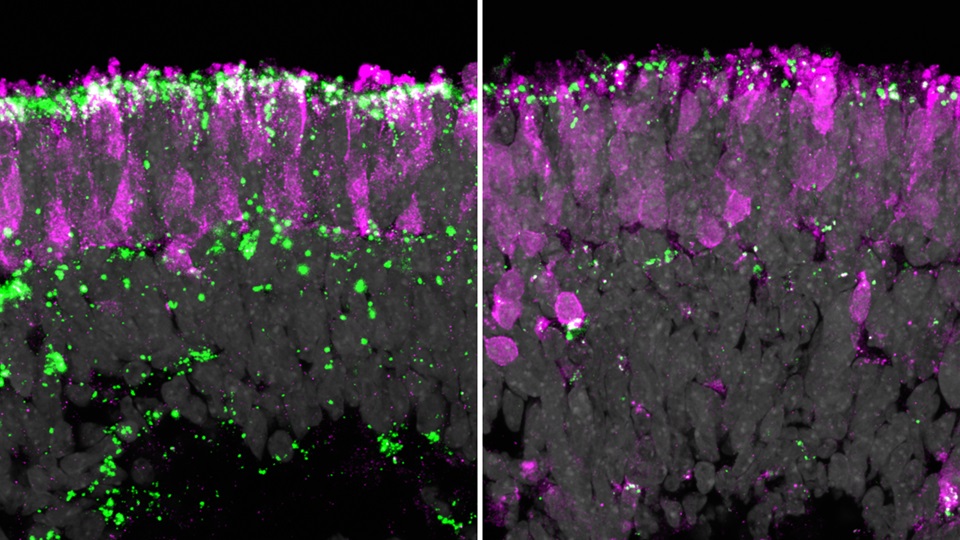
»Unsere Netzhaut‑Organoide zeigen, dass wir Erkrankungen des Auges auf diese Weise realistisch im Labor nachbilden und die Wirksamkeit des gentherapeutischen Ansatzes erfolgreich daran erproben können. Und das in einer für den Menschen aussagekräftigen Art und Weise und ohne dabei auf Tierversuche zurückgreifen zu müssen«
Forschungsgruppenleiter Dr. rer. nat. Kevin Achberger
Voraussetzung für klinische Studie
Diese präklinischen Ergebnisse ermöglichten die Genehmigung zum Start einer klinischen Studie für Kinder mit CLN2-bedingten Netzhautschäden in England – und das ohne eine vorherige Testung der Wirksamkeit der Gentherapie an Tiermodellen. Die ersten Zwischenergebnisse der klinischen Studie TTX-381 zeigen erste Anzeichen, dass durch die Gentherapie eine Stabilisierung und sogar Verbesserung des Sehvermögens bei Patientinnen und Patienten erzielt wurde.
Gentherapie für weitere neurologische Krankheitsaspekte
Parallel zur Netzhauttherapie arbeitet Tern Therapeutics an einer zweiten Gentherapie (TTX-181), die direkt im Gehirn der Betroffenen wirken soll. Ziel ist es, nicht nur die Erblindung zu verhindern, sondern den neurologischen Krankheitsverlauf zu verlangsamen oder sogar zu stoppen und die Lebenserwartung deutlich zu verlängern. »Einer der größten Ängste von Familien, deren Kinder an CLN2 erkrankt sind, ist der Verlust des Sehvermögens. Deshalb war es so wichtig, uns zunächst auf die Behandlung des Sehverlusts bei CLN2 zu konzentrieren. Unserer Therapie hat das Potenzial, die Lebensqualität der kleinen Patientinnen und Patienten erheblich zu verbessern. Wir sind uns jedoch der vielen anderen Herausforderungen bewusst, denen diese Kinder und ihre Familien gegenüberstehen, und entwickeln daher auch Gentherapien zur Behandlung anderer Aspekte der CLN2-Erkrankung«, erklärt Alex M. Bailey, CEO von Tern Therapeutics.
Paradigmenwechsel in der Medikamentenforschung
Diese Zusammenarbeit repräsentiert einen Durchbruch: Sie zählt zu den ersten Projekten, bei denen Gentherapien ohne Wirksamkeitstests an Tieren zur klinischen Erprobung gelangen konnten. Das Modell der Retina-Organoide in Kombination mit der Organ-on-Chip-Technologie eröffnet neue Möglichkeiten für die Medikamentenentwicklung durch die zum Teil realistische Nachbildung von menschlichen Erkrankungen. Ein zusätzlicher Vorteil liegt in der potenziellen Verkürzung der Zeitspanne zwischen Medikamentenentwicklung und Behandlung der Patientinnen und Patienten, dies ist insbesondere für Betroffene mit seltenen Krankheiten von entscheidender Bedeutung. Entgegen ihrer Bezeichnung leiden weltweit insgesamt rund 400 Millionen Menschen an einer seltenen Krankheit.
Quelle: Universitätsklinikum Tübingen (zur Pressemitteilung)
Originalpublikation: Corti S, Kim KH, Chen T, Botezatu A, Cora V, Ma K, et al. Recreating pathophysiology of CLN2 disease and demonstrating reversion by TPP1 gene therapy in hiPSC-derived retinal organoids and retina-on-chip. Cell Rep Med 2025; 6: 102244. https://doi.org/10.1016/j.xcrm.2025.102244
Bilderquelle: © Ramona Heim – stock.adobe.com; Symbolbild


